Integrating Data to Predict Functions for Gaps in Metabolic Models
Authors:
William C. Nelson1* ([email protected]), Jason McDermott1,2, David Geller-McGrath3, Christine Chang1,Jeremy Jacobsen1, Tara Nitka1, Travis Wheeler4, Robert Egbert1 (PI)
Institutions:
Goals
The Persistence Control Science Focus Area (PerCon SFA) at Pacific Northwest National Laboratory seeks to understand plant-microbiome interactions in bioenergy crops to establish plant growth–promoting microbiomes that are contained to the rhizosphere of a target plant. This vision requires the discovery of exudate catabolism pathways from plant roots, the elimination of genes that support fitness in bulk soil environments without decreasing rhizosphere fitness, and the engineering of rhizosphere niche occupation traits in phylogenetically distant bacteria. The team anticipates the impacts of these efforts will be to increase understanding of plant-microbe interactions and to extend high-throughput systems and synthetic biology tools to non-model microbes.
Abstract
Metabolic modeling in bacterial genomes or metagenomes depends on several critical steps. Assigning functions to predicted genes and populating known metabolic pathways and modules with annotated genes precede the assembly a draft model. Typically, the limitations of functional annotation reveal critical reaction steps, or gaps, in a draft model for which no gene was identified. Further, comparison of the draft model against experimentally derived phenotypic data, such as cell growth experiments, can identify additional gaps or inconsistencies in the model. Gaps are typically filled either by simply assuming the that the function exists with no specific gene assignment to the step or by relaxing thresholds and computationally or manually searching original annotations for lower confidence or less specific annotations. There are multiple significant impediments to the metabolic modeling and gap-filling process: (1) incomplete genome information, including for metagenome-assembled genomes (MAGs); (2) low-confidence annotations from bacteria that are evolutionarily distant from well-studied organisms; and (3) the large pool of protein families that have either no functional assignment or a non-specific function.
This team has developed two tools to overcome these deficiencies. MetaPathPredict (Geller-McGrath et al. 2022) is a deep learning framework for prediction of complete metabolic modules in genome-scale models from incomplete genome data. Employing a gradient-boosted trees [XGBoost (Chen and Guestrin 2016)] and neural network stacked ensemble classification framework, MetaPathPredict can accurately predict the presence of metabolic modules with as much as 60 to 70% of the genome missing by learning from complete genomes.
The team also developed Snekmer (Chang et al. 2023), a generalized computational framework to model protein function families using sequence recoding into reduced-complexity amino acid alphabets and a k-mer based approach. Snekmer can be used to rapidly develop models for novel protein families and to screen genomes or metagenomes to evaluate the distribution of the family across organisms or environments. Using these tools to fill gaps will improve researchers’ ability to develop metabolic models from limited genome data, including MAGs. Team members have successfully integrated Snekmer as a KBase app, and next plan to integrate MetaPathPredict. Both tools will function with the new OMics-Enabled Global Gapfilling (OMEGGA) app in KBase to allow iterative model building, gap filling, and model refinement.
Image
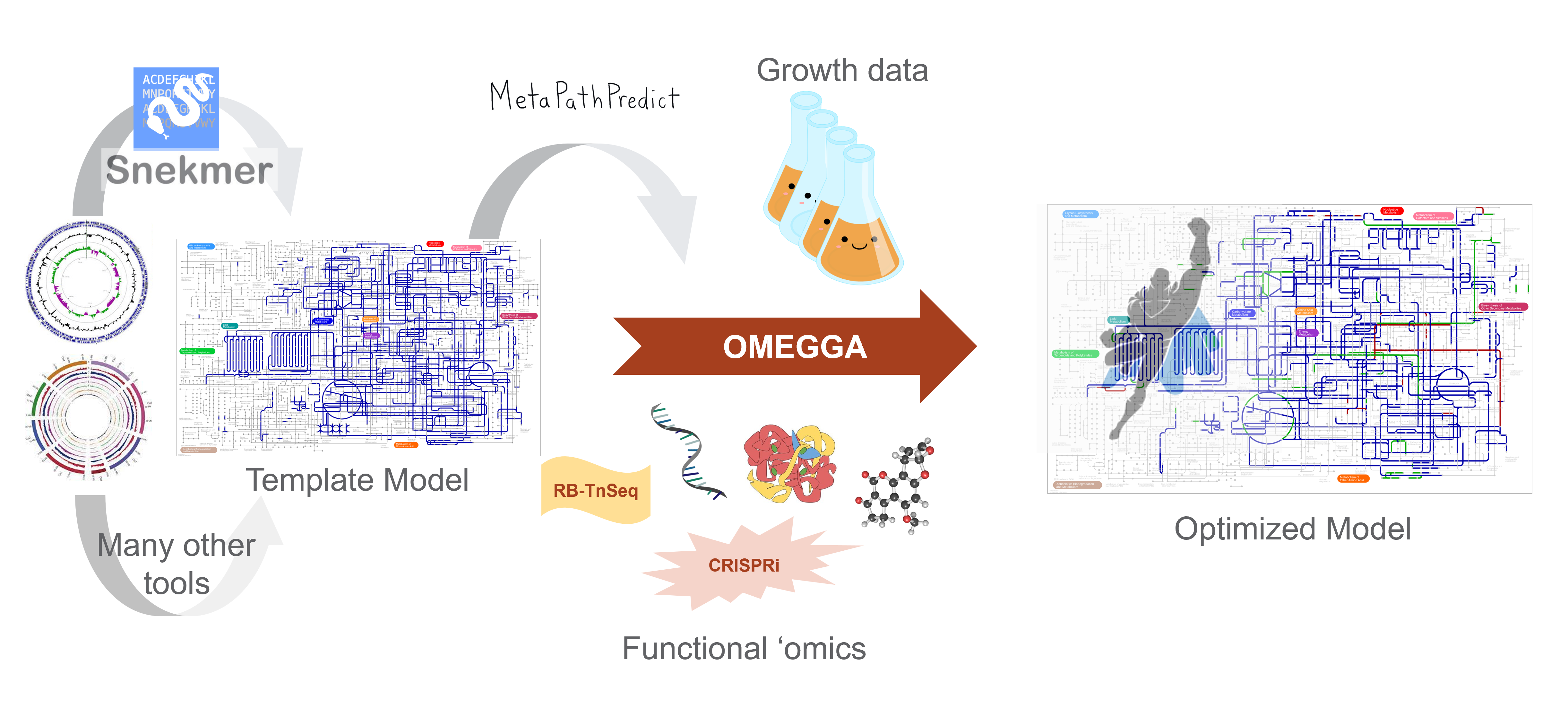
Metabolic Model. Machine learning is the basis of many new tools for processing a wide and diverse range of experimental data for the purpose of building (Snekmer), improving (MetaPathPredict), and parameterizing (OMics-Enabled Global Gapfilling app) metabolic network models to optimize their accuracy and biological relevancy. [Courtesy Pacific Northwest National Laboratory]
References
Chang, C. H., et al. 2023. “Snekmer: A Scalable Pipeline for Protein Sequence Fingerprinting Based on Amino Acid Recoding,” Bioinformatics Advances 3(1), vbad005. DOI:10.1093/bioadv/vbad005.
Chen, T., and C. Guestrin. 2016. “XGBoost: A Scalable Tree Boosting System.” KDD ’16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 785–95. DOI:10.1145/2939672.2939785.
Geller-McGrath, D., et al. 2022. Preprint. “MetaPathPredict: A Machine Learning-Based Tool for Predicting Metabolic Modules in Incomplete Bacterial Genomes,” bioRxiv. DOI:10.1101/2022.12.21.521254.
Funding Information
This work was supported by the DOE BER program and is a contribution of the Science Focus Area Persistence Control of Engineered Functions in Complex Soil Microbiomes, the DOE/BER “Improved Protein Annotation in KBase Using Machine Learning, Multiomics Data Integration, and Structural Prediction” project, the DOE/BER “Machine-Learning Approaches for Integrating Multiomics Data to Expand Microbiome Annotation” project, National Science Foundation (1856556), the Defense Threat Reduction Agency, under Interagency Agreement (DTRA13081–37739), and the Proxy Applications for Converged Workloads (PACER) project, a component of the Laboratory Directed Research and Development program at Pacific Northwest National Laboratory. Pacific Northwest National Laboratory is operated for DOE by Battelle Memorial Institute under contract DE-AC05-76RL01830.