Provide the basic science needed to convert renewable biomass to a range of fuels, chemicals, and other bioproducts in support of a burgeoning bioeconomy.
The Genomic Science Program’s (GSP) bioenergy research efforts seeks to
- Gain a genome-level understanding of plant metabolism, physiology, and growth to develop new bioenergy feedstocks with traits tailored for bioenergy and bioproduct development
- Develop an understanding of microbial and fungal metabolism necessary to design new strains, communities, or enzymes capable of converting plant biomass components into fuels, chemicals, and bioproducts.
- Understand the genomic properties of plants, microbes, and their interactions to enable the development of new approaches that improve the efficacy of bioenergy crop production on marginal lands with few or no agricultural inputs, while minimizing ecological impacts under changing environmental conditions.
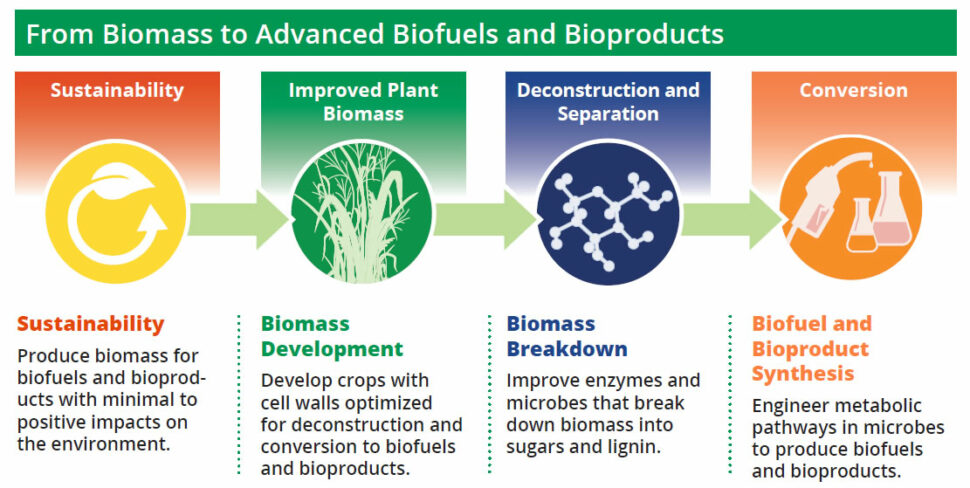
Many recent efforts have been performed within the four DOE Bioenergy Research Centers (BRCs). These large, interdisciplinary BRC teams have provided key basic science insights needed to spur broader development of biofuels and bioproducts from lignocellulosic plant biomass. Continued research will seek to expand this body of knowledge to extend the range of biofuels and bioproducts that can be produced from biomass feedstocks.
Research Areas

Plant Genomics
Gain a genome-level understanding of plant metabolism, physiology, and growth to develop new bioenergy feedstocks with traits tailored for bioenergy and bioproduct development.
Microbial Conversion
Develop an understanding of microbial and fungal metabolism necessary to design new strains, communities, or enzymes capable of converting plant biomass components into fuels, chemicals, and bioproducts.

Sustainability
Understand the genomic properties of plants, microbes, and their interactions to enable the development of new approaches that improve the efficacy of bioenergy crop production on marginal lands with few or no agricultural inputs, while minimizing ecological impacts under changing environmental conditions.
GSP plans to maintain investments in core strengths related to plant genomics and the development of dedicated bioenergy crops, deconstruction of plant biomass, microbial conversion of biomass to fuels and products, and the sustainability of bioenergy crop production. GSP will support these efforts through awards to large, integrative teams, such as the BRCs, smaller team research projects, and single investigators. Additionally, the program will seek to encourage greater integration among the current BRCs toward common goals, objectives, and milestones that chart a path for the next decade of bioenergy research.
Today’s liquid fuels and many of the chemical products and materials that support modern society are primarily derived from petroleum and other fossil resources. Producing fuels, chemicals, and bioproducts (including materials) from renewable plant biomass could reduce our dependence on fossil resources and pave the way toward a more sustainable bioeconomy.
Bioenergy Research Centers
Since 2007, the Genomic Science program has supported the Bioenergy Research Center (BRC) program, whose mission is to break down the barriers to actualizing a domestic bioenergy industry. The centers—each led by a DOE national laboratory or top university—take distinctive approaches toward the common goal of accelerating the pathway to improving and scaling up advanced biofuel and bioproduct production processes.
- Center for Advanced Bioenergy and Bioproducts Innovation (CABBI), led by the University of Illinois at Urbana-Champaign. CABBI is integrating recent advances in agronomics, genomics, biosystems design, and computational biology to increase the value of energy crops, using a “plants as factories” approach to grow fuels and chemicals in plant stems and an automated foundry to convert biomass into valuable chemicals that are ecologically and economically sustainable.
- Center for Bioenergy Innovation (CBI), led by Oak Ridge National Laboratory. CBI is accelerating the domestication of bioenergy-relevant plants and microbes to enable high-impact, value-added coproduct development at multiple points in the bioenergy supply chain.
- Great Lakes Bioenergy Research Center (GLBRC), led by the University of Wisconsin—Madison in partnership with Michigan State University. GLBRC is developing the science and technological advances to ensure sustainability at each step in the process of creating biofuels and bioproducts from lignocellulose.
- Joint BioEnergy Institute (JBEI), led by DOE’s Lawrence Berkeley National Laboratory. JBEI is using the latest tools in molecular biology, chemical engineering, and computational and robotics technologies to transform biomass into biofuels and bioproducts.